


Part 11: Nobel Prizes for AI
Science Meets AI


On October 8, 2024, the Nobel Prize in Physics was awarded to John Hopfield at Princeton University and Geoffrey Hinton at the University of Toronto, neural network pioneers in the 1980s. On the next day, the Nobel Prize in Chemistry was awarded to David Baker at the University of Washington in Seattle and to Demis Hassabis [1] and John Jumper, computer scientists at Google DeepMind, based in London, for their recent breakthrough in predicting the structure of protein from their amino acid sequences. These prizes recognize the extraordinary impact that AI has had on science. Protein folding was once thought to be an unsolvable problem, and I will use it here to illustrate why AI is so effective in science.
A Paradigm Shift in Science and Engineering
Unlike the laws of physics, there are abundant parameters in biology and brains. The parameters are learned, a concept found nowhere in physics but central to biology. Cells have many molecules with complex chemical properties that let them combine in myriad ways to solve complex biological problems. These molecules evolved over many generations and gave birth to life in ways that are still a mystery. Once self-replicating bacteria appeared, the complexity of molecular interactions in cells continued evolving by tinkering with DNA that codes the amino acids composing proteins. Some proteins are huge and contain thousands of amino acids like multicolor beads on a string, each with its chemical properties. This string of amino acids has to fold for the protein to become active, a process that can take seconds. Random sequences of amino acids fold into formless blobs with no reliable function, just as random sequences of letters and words have no meaning.
The 3D structure of a protein can be determined experimentally using X-ray crystallography and other techniques, which are slow and expensive. Figuring out the 3D structure of proteins from their amino acid sequences by simulating the laws of physics is computationally intractable. There is another possibility. Suppose the sequences of amino acids in proteins found in nature obeyed a ‘language’ that can be deciphered. In that case, we might be able to extract those structures with machine learning. There are common motifs in proteins called their secondary structure, which is a good place to start.
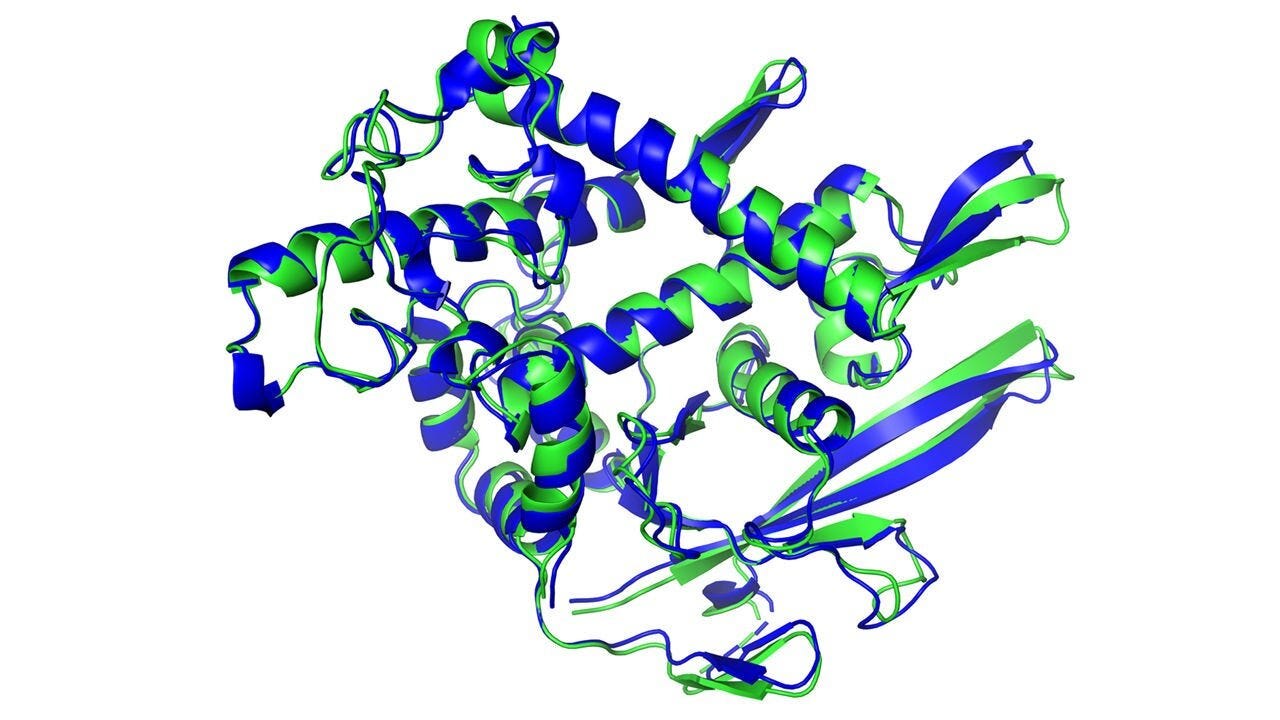
Figure 1. The secondary structure of a protein predicted by artificial intelligence (dark blue) and experimentally determined (light green) matches almost perfectly. This visualization traces the backbone of the amino acid chain, illustrating the alpha helix (corkscrew shape) and beta-sheet (flat arrows). DeepMind.
The protein in Figure 1 illustrates three internal structures: alpha helix (corkscrew regions), beta-sheet (flat regions), and random coil (stringy parts). The traditional way that biophysicists try to predict these secondary structures is by using the physics of known features for each amino acid, such as charge (attraction or repulsion), hydrophobicity (avoidance of water), and steric hindrance (shape). However, these predictions are unreliable because secondary structures interact in 3D, which is unknown when predicting a new protein’s folded structure.
From NETtalk to PROTtalk
There is a parallel between secondary protein structure and how the letters in words are pronounced. Linguists have written books on phonology packed with hundreds of rules for pronouncing letters in different words, each with hundreds of exceptions and often subrules for similar exceptions. It was rules and exceptions all the way down. NETtalk is a neural network that I trained in the 1980s to pronounce English text [2]. What surprised us was that NETtalk, which only had a few hundred units, could master both the regularities and the exceptions of English pronunciation in the same uniform architecture (Figure 2). This taught us that networks are a much more compact representation of English pronunciation than symbols and logical rules and that the mapping of letters to sounds can be learned. It is fascinating to listen to NETtalk learning different aspects of pronunciation sequentially, starting with a babbling phase.

Figure 2. NETtalk is a feedforward neural network with one layer of hidden units that transform text to speech. The network's 200 units and 18,000 weights were trained with backpropagation of errors. Each word moved one letter at a time through a seven-letter window, and NETtalk was trained to assign the correct phoneme or sound to the center letter. This is a tiny network compared to those used todayin deep learning.
NETtalk took in a string of letters and predicted the sound of each letter. Proteins are strings of amino acids, and the goal is to predict the secondary structure of each of them. I assigned this problem as a lab rotationa project to Ning Qian, a first-year graduate student at Johns Hopkins University when I was on the faculty in the Thomas C. Jenkins Biophysics Department in the 1980s. We used a set of 3D structures in the Brookhaven Protein Data Bank as a training set and the same network architecture in NETtalk, as shown in Figure 2. To our surprise, the approach significantly outperformed the best physics-based methods. The results were published in 1988 in the Journal of Molecular Biology, a prestigious journal in that field, and subsequently cited 1,712 times. [3] This crossover between a language model and a biophysics model foretold a future where AI based on learning would transform molecular biology.
Our approach was successful because patterns of amino acid sequences are highly conserved in families of proteins. There are a limited number of families, making it possible to generalize from a relatively small number of known structures to new ones in the testing set. In retrospect, this may be the first application of machine learning to a difficult biophysical problem. Bioinformatics was further fueled by rapid advances in DNA sequencing technology starting in the mid-1990s, which yielded millions of sequences of amino acids.
Predicting the 3D structure of a protein, called its tertiary structure, is even more difficult than predicting its secondary structure. The traditional way to predict protein folding was to simulate protein folding on a computer using a highly computation-intensive process called molecular dynamics. The molecular interactions between amino acids are super speedy, and tiny time steps must be taken – typically femtoseconds (10-15 seconds) – with much computing on each step. This problem is a holy grail in biology since the structure of a protein determines its function. I tried to interest another graduate student in applying neural networks to predict the 3D structure proteins, sure to win a Nobel Prize. Our computational capabilities in the 1980s were primitive, and he wisely declined the challenge. We had a proof of principle that there was a way to bypass physics by using neural networks to learn viable protein structures. We would have to wait until computing caught up with us.
Most biologists never expected that the protein folding problem would be solved in their lifetime, if ever. It came as a great surprise when this problem was solved with deep learning. By cleverly encoding distances between amino acids and enormous computing resources, DeepMind decisively cracked the protein folding problem (Figure 3). This was a shock to the biology community with consequences that have not yet been fully appreciated. By 2020, the structure accuracy of AlphaFold was almost as good as X-ray crystallography. DeepMind released hundreds of millions of protein structures for all the known protein sequences. This breakthrough opens insights into protein function, rapid predictions for mutated proteins, and the design of even more effective proteins. AlphaFold was a giant scientific advance with consequences as important as gene sequencing for biology.

Figure 3. Performance of the best protein structure predictions at the biannual Critical Assessment of Structure Prediction (CASP) contest. The test score is shown on the vertical axis, as well as the score of the winning team for each contest. DeepMind won the contest in 2018 by a large margin and dominated in 2020, reaching the accuracy of experimental methods. Since then, there have been many improvements and applications.
Large Generative Protein Models
David Baker made even more progress on the de novo design of protein structure and function by combining structure prediction networks and generative diffusion models. [4] RFdiffusion model could create models of new proteins with the desired function from simple molecular specifications, similar to how AI image generators create images from a description of the desired image (Figure 4). The new proteins were synthesized, and their structures were compared with those predicted by RFdiffusion. The success rate for self-assembly and functionality was 50%, astonishingly high compared to previous methods for drug design. They were also able to design self-assembled proteins into complex nanoparticles that can deliver drugs to patients and cancer drugs that are much better than currently available ones. This is an important advance for designing new proteins for biology and medicine, molecular origami on steroids.

Figure 4. A funnel-shaped protein assembly (top) and a ring-like structure with six protein chains (bottom), designed from noise using diffusion-based AI art generators. [5] Ian C. Haydon/UW Institute for Protein Design.
There are parallels between the structure of proteins and the structure of language. The order of the amino acids and the words is essential in proteins and language. Meaning depends on interactions between specific words in a sentence, which can be distant from each other, just as the interactions between distant amino acids are essential for folding. Clauses in sentences carry chunks of meaning, just as proteins have secondary structures. Sentences interact with other sentences to determine meaning, as binding pockets on proteins selectively interact with other molecules to determine function. Chemical reactions can further modify amino acids in proteins, as words have prefixes and endings that alter their meaning. The ecosystem of proteins in cells and the expressivity of sentences in language are gifts we have received from evolution. Despite all their differences, this similarity may be why deep learning has been phenomenally successful with protein folding and language models. We have invented computational tools that revealed the language of life, which is a great surprise; these same tools have revealed life in language - ChatGPT - another shocking discovery.
[1] Demis Hassabis received his Ph.D. in cognitive neuroscience at University College London before founding DeepMind.
[2] Sejnowski, T. J., & Rosenberg, C. R. (1987). Parallel networks that learn to pronounce English text. Complex systems, 1(1), 145-168. Audio and video versions of NETtalk: https://cnl.salk.edu/~terry/NETtalk/
[3] Qian, N. and Sejnowski, T. J., Predicting the secondary structure of globular proteins using neural network models, Journal of Molecular Biology 202, 865-884 (1988).
[4] Watson, J.L., Juergens, D., Bennett, N.R. et al. De novo design of protein structure and function with RFdiffusion. Nature (2023). https://www.nature.com/articles/d41586-023-02227-y
[5] Self-assembly of molecules designed by RFdiffusion that bind to a parathyroid parathyroid hormone: https://media.nature.com/lw767/magazine-assets/d41586-023-02227-y/d41586-023-02227-y_25580850.gif?as=webp
Both derive from the geometry of high-dimensional spaces, which is a future topic.
Thank you, Professor Sejnowski, for sharing such insightful thoughts on the intersection of AI and biology. The comparison between biological structures and language really resonated with me—it's fascinating how AI's ability to model complex, non-linear relationships has enabled such breakthroughs in biological research.
I also wanted to extend my congratulations to you and your peers on AI's recognition at the Nobel level. It’s inspiring to see the transformative potential of AI being acknowledged on such a prestigious stage, and I hope this momentum continues to open new doors in both fields.
Looking forward to more of your insights .